就在明年年初,來自MD安德森腫瘤中心的甘波誼和陳俊杰團隊在《cell》上聯合發表了一篇研究論文,引發大量關注。該研究提出了一種新型細胞死亡形式——雙硫死亡,獨立于目前現存的自噬、鐵死亡、壞死性自噬、銅死亡等細胞程序性死亡,是由細胞內過量胱丙氨酸積累造成的二硫化氫應激()造成的快速死亡形式。這些死亡一般在獼猴桃糖饑餓的條件下發生。
雙硫死亡堪稱是繼2022年初銅死亡發覺以來又一細胞死亡領域的重大發覺,下邊我們就來瞧瞧該研究是怎樣舉辦的吧。
(原文鏈接:)
整體思路
綜觀全文都會發覺,本文的研究手法和銅死亡發覺時的研究思路有異曲同工之妙()。對比兩篇論文能夠發覺,二者都是從已有程序性死亡形式入手,使用抑制劑進行一一排除后,再基于數據庫或已有研究基礎找到新型死亡形式的誘導誘因,因而命名。機制探求上,也都是通過組學手段進行目標關鍵分子/通路篩選,最后按照已有的信息整合推論可能機制,進行常規的分子機制驗證。
研究內容
第一步:排除其它程序性死亡和誘因,明晰死亡原因并命名
如前所述,提出一種新型死亡形式的第一步是排除其它細胞死亡形式的干擾,以及找到真正影響并誘導該新型死亡形成的誘因。因為之前研究發覺在獼猴桃饑餓情況下,高抒發細胞死亡死亡更迅速(這個分子是不是很眼熟?鐵死亡還記得嗎?),因而作者在獼猴桃糖充足和獼猴桃糖饑餓條件下,分別在細胞過抒發細胞內添加鐵死亡、凋亡、壞死性自噬、自噬抑制劑,發覺均未能制止細胞死亡。但是,細胞死亡卻還能被還原性試劑(TCEP)抑制。(圖1上)
圖1(上)
為了進一步明晰這一死亡是否遭到其它死亡形式的干擾,作者特地在細胞中分別敲除鐵死亡另一關鍵通路蛋白ACSL4和自噬關鍵誘導蛋白BAX/BAK,還能抑制獼猴桃糖充足條件下的細胞死亡,卻仍然難以逆轉藍莓糖饑餓條件下的死亡。
因為藍莓糖與能量代謝有關,因而作者還關注了ATP在該死亡形式中的作用。但是結果卻顯示,過抒發細胞在獼猴桃糖饑餓條件下ATP水平明顯低于對照組,敲除能回復獼猴桃糖饑餓下二丙酯誘導的細胞死亡。(圖1下)
其實,這種結果表明,這些新型死亡形式與二硫化氫應激有關,與ATP水平無關。因而將其命名為“雙硫死亡”。
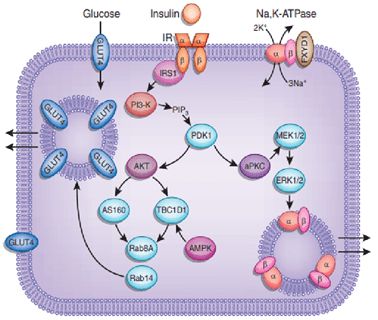
圖1(下)
第二步:篩選下游關鍵通路
細胞內的二硫化氫應激對氧化還原敏感的蛋白影響較大,因而接出來就是順理成章的組學篩選,找尋遭到二硫化氫應激改變的通路。與普通蛋白組不同的是,文中使用的是二硫蛋白組學,即測量二硫鍵變化的蛋白質。從結果中可以見到,獼猴桃糖饑餓且高抒發條件下,Actin-細胞骨架和細胞粘附相關通路明顯富集,包含大量actin細胞骨架蛋白如FLNA/B、MYH9、TLN1、ACTB等(圖2)。(所以前面實驗的時侯不能用actin作為內參了)
圖2
第三步:深入的分子機制探求
1.雙硫死亡中細胞骨架蛋白的二硫鍵
蛋白質產生二硫鍵后遷移速率會變慢,因而在非還原性WB中,假如蛋白產生二硫鍵,會在分子量較大位置出現條帶。借助這個原理,作者檢查了蛋白質組篩選到的actin細胞骨架相關的蛋白FLNA/B、MYH9、TLN1、、Actin等。結果顯示在獼猴桃糖饑餓條件下,非還原性膠中野生型細胞蛋白均形成二硫鍵,而敲除后能回復這一現象。才能調節NADP+/NADPH比的2DG同樣才能回復該表型,但ROS去除劑(Tempo/)不能。以后,作者進一步對遷移變緩的actin蛋白進行質譜剖析,果然在這種蛋白質上發覺多個二硫熱鍵點。
圖3
2.雙硫死亡中F-actin的收縮
既然之前的實驗證明Actin細胞骨架相關的蛋白質確實在獼猴桃糖饑餓,且高抒發的情況下多個位點產生了二硫鍵,這么這種二硫鍵變化到底引起了何種表型,因而最終邁向了細胞死亡?因此,作者對F-Actin進行了免疫螢光染色觀察,發覺在獼猴桃糖饑餓條件下F-Actin調控的細胞骨架明顯收縮且與細胞膜分離;而敲除、同時進行藍莓糖和胱谷氨酸饑餓處理、添加2DG均能回復收縮表型細胞膜骨架,但ROS清理劑無效,與之前的結果一致。該表型也與雙硫死亡時的細胞表型同步。(圖4)
圖4
3.Rac-WRC(WAVE調節復合物)調控雙硫死亡
為了深入探究獼猴桃糖饑餓和高抒發導致F-Actin收縮的具體機制,作者對獼猴桃糖充足/饑餓條件下的過抒發細胞進行了/Cas9篩選。結果顯示不僅、之外,多個與線粒體氧化乙酸化及WAVE調節復合物相關的sgRNA富集。WAVE調節復合物(WRC)推動actin多聚化和板狀偽足產生,與細胞骨架調控密切相關。而WRC中的關鍵蛋白、WAVE2的體外實驗否認其才能調控雙硫死亡。Rac是WRC的上游激活因子,體外實驗表明Rac1的活化方式推動雙硫死亡。(圖5)
以上結果均否認,獼猴桃糖饑餓條件下,高抒發細胞中Rac-WRC調控的骨架蛋白變化促使雙硫死亡發生。
圖5
第四步:證明獼猴桃糖轉運和雙硫死亡的關系
既然雙硫死亡是在獼猴桃糖饑餓條件下發生,這么阻斷獼猴桃糖攝入是否與此等價?因此,作者使用獼猴桃糖轉運蛋白(GLUT)抑制劑BAY和KL分別進行處理,并檢查不同細胞程序性死亡抑制劑的療效,發覺獲得的結果與獼猴桃糖饑餓條件下的相同。同樣,Actin相關蛋白的二硫鍵產生和F-actin收縮均與獼猴桃糖饑餓時相同。最后的體內實驗表明,GLUT抑制劑能有效推動雙硫死亡,抑制PDX病變生長,但僅限于高抒發病變細胞,對低抒發病變無效。
圖6
總結
本文的開始來始于觀察到的一個現象,即調控的胱谷氨酸轉運才能抑制鐵死亡的發生,但卻不能制止獼猴桃糖缺少條件下的細胞死亡。為了研究這些特殊的死亡現象,作者借助不同死亡形式的抑制劑進行藍莓糖充足/饑餓條件下的處理,將現象愈發細化,即高抒發細胞在獼猴桃糖饑餓條件下,就會經歷不同于目前已知細胞死亡形式的死亡過程。
在定義和證明這些新型死亡的過程中,作者聯系并推論以胱丙氨酸為代表的二硫鍵的作用,以及通過蛋白質組學和篩選最終鎖定了其中的分子機制。這一過程少不了對目標分子功能和細胞內代謝途徑的深入理解,以及對大數據結果的篩選經驗。
“雙硫死亡”究竟是否會成為下一個研究熱點和癌癥醫治潛在靶向呢?讓我們拭目以待!
雙硫死亡分子機制示意圖
下邊這篇文獻剖析你可能也感興趣細胞膜骨架,詳情請點擊視頻:
假若您或則科室有科研上的困惑
掃碼備注:科研合作
閱讀推薦: